
A Quarter-Century Quest to Treat a Rare Disease
NIDCR research on FD/MAS offers hope, answers, and therapies
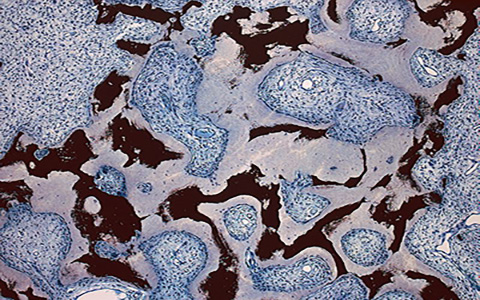
One day in early 1993, NIDCR scientist Pamela Robey, Ph.D., received a box full of bone samples from an endocrinologist colleague. The bone had been taken from patients with a rare disease that causes early puberty, dark patches on the skin, and fragile, misshapen bones, among other symptoms. For many, the disorder, called fibrous dysplasia/McCune-Albright syndrome (FD/MAS), can be extremely painful, disabling, and impair quality of life.
"I took a look at the samples under a microscope and was taken completely by surprise," said Dr. Robey, a skeletal stem cell biologist. "Instead of blood-rich marrow with associated bone, parts of the marrow cavity were completely filled with poorly mineralized, disorganized bone, and fibrotic tissue. It seemed like the bone stem cells, normally inconspicuous in the marrow, had gone absolutely haywire."
The misbehaving bone cells intrigued Dr. Robey, who was interested in understanding bone development and the ways it can go awry to cause human disease. If she could figure out what had gone wrong with the patients’ bone stem cells, there might be a way prevent it from happening in the first place. Little did Dr. Robey know that by opening the box, she would spark a journey that, so far, has been going on for 25 years at NIDCR. Its aim is to better understand and treat FD/MAS.
Cracking Down on the Rare Disease
The colleague who brought the bone samples was physician-scientist Allen Spiegel, M.D., then the scientific director of the National Institute of Diabetes and Digestive and Kidney Diseases. Dr. Spiegel was studying diseases that arise from mutations in genes that code for a family of proteins called G proteins. These proteins are found in cells throughout the body and are important for the proper functioning of a wide range of biological processes, including those of the immune, nervous, and endocrine systems.
A couple years earlier, Dr. Spiegel had found that a nonhereditary mutation of the gene that codes for one member of this protein family — Gαs — appeared to be responsible for the skin and hormonal effects of FD/MAS. But he needed Dr. Robey’s expertise as a bone stem cell biologist to answer the question of how the mutation might also cause the disease’s bone defects.
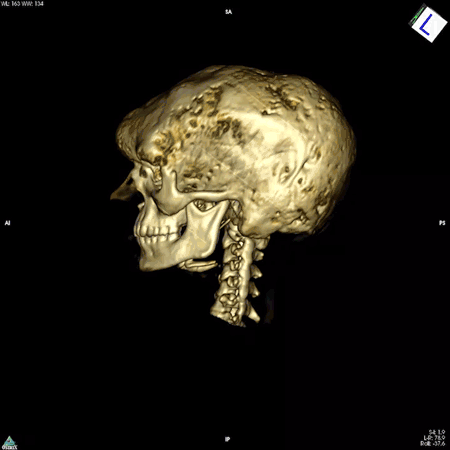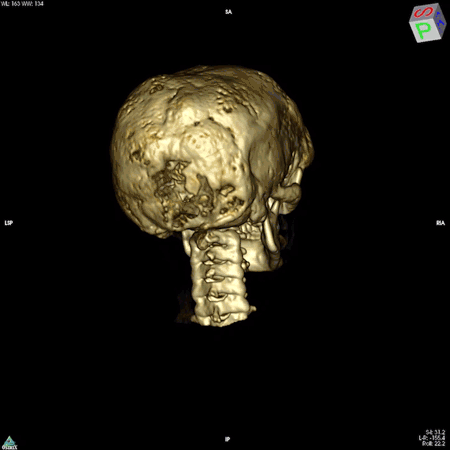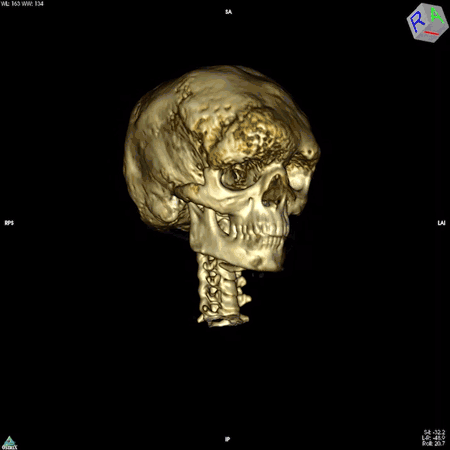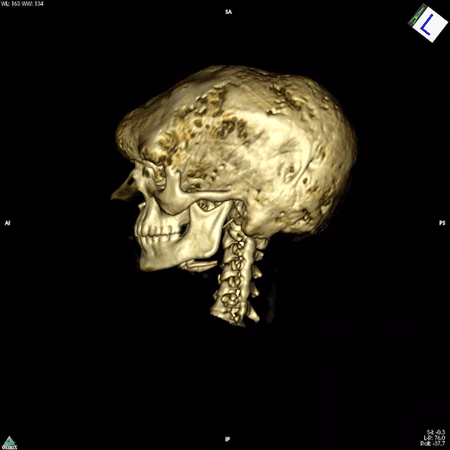
The answer turned out to be multifaceted. Dr. Robey, along with colleagues in Italy, discovered that the mutation impairs the ability of skeletal stem cells to develop into mature bone cells. In addition, these mutation-carrying cells multiply rapidly and form bone tissue that doesn’t harden like it should, resulting in weak regions throughout the bone. These faulty patches, or lesions, cause bones to bow and break easily and to expand abnormally. The bones of the face and skull are among the most affected. The lesions can also press up against organs and nerves, impairing functions like vision, hearing, and breathing. While there are drugs to manage the hormonal symptoms of the disease, none exist to slow or curb the development of bone lesions.
A Vision for Patient-Centered Research
Dr. Robey realized that with NIDCR’s expertise in bone biology, she and her colleagues might have a shot at finding treatments for the skeletal aspects of the disease. To that end, in 1998, Dr. Robey, along with NIDCR investigator and endocrinologist Michael Collins, M.D., who was then a clinical fellow, launched a research project, called a natural history study, to document the nature and course of the disease. A quarter of a century later, the work is still ongoing and has enrolled more than 300 patients ages 1 to 102. This longstanding engagement with the patient community is one of the key drivers of FD/MAS research.
“At the end of the day, it’s the patients who we serve,” said Dr. Collins, who eventually took the lead on the study, serving as principal investigator until 2016. “Talking and listening to these patients is how we really learn what their problems are and what issues need to be addressed; it really frames the important questions to answer.”
In the natural history study, clinicians inspect patients from head to toe by taking bone scans, collecting tissue samples, and documenting every characteristic of the disease.
The work has revealed that each person with FD/MAS experiences the disease differently. Some people have severe disease throughout their bodies. Others have mild symptoms that are confined to a specific body system. This variability occurs because the Gαs mutation arises early in embryonic development. The mutation-containing cells spread unevenly into regions of the embryo that eventually become different body parts. Each patient has a different number and distribution of mutation-containing cells, making their condition and its severity unique.
Because FD/MAS is so highly variable, there is no one-size-fits-all treatment for the disease. Dr. Collins and NIDCR investigator and pediatric endocrinologist Alison Boyce, M.D., who has led the natural history study since 2016, addressed this issue by creating a way to personalize the care of each patient. Their clinical decision-making algorithm allows clinicians to input a patient’s symptoms and receive a treatment plan tailored to that patient’s unique situation.
NIDCR researchers have also helped clear up uncertainty among clinicians around the need for a potentially risky surgery to the optic nerve. In many cases, fibrous dysplasia lesions constrict the optic nerve, which had been thought to account for the vision loss often experienced by patients. For this reason, some clinicians recommended preventative surgery for patients whose nerves were constricted but who had not yet experienced vision problems. However, even in patients with vision loss, surgery to decompress the nerve is not always effective, and can even cause blindness. Clinicians in the field questioned the need for such a risky surgery in patients whose vision has not yet been affected. A team led by Dr. Collins and NIDCR Clinical Director Janice Lee, D.D.S., M.D., found in 2002 that among 38 patients whose computed tomography (CT) scans showed compression of the optic nerve, 95% had normal vision. The finding suggested that constriction alone does not cause vision loss and that the risks of preventative surgery may outweigh the benefits for patients whose vision is normal.
“Much of the research in clinical management of the craniofacial aspects of FD/MAS has been conducted by the NIDCR clinical team, thanks in large part to clinicians in specialties such as dentistry, maxillofacial surgery, otolaryngology, neuro-ophthalmology and endocrinology working side by side to understand this complex disease,” said Dr. Lee.
Search for Bone Therapy
The information NIDCR scientists have learned from the patients has also spurred studies in cells and animals to further tease apart the factors that contribute to the disease and to test potential therapies. For example, research using cells from patients helped point to a potential therapy: an FDA-approved osteoporosis drug called denosumab.
In lab experiments using bone cells from patients with FD/MAS, the drug blocked a protein responsible for excessive bone turnover, a hallmark of the disease. Bone turnover is the process by which old bone is continually replaced with new bone.
"FD/MAS is a challenging disease to study, because it progresses over the years," said Dr. Boyce. "It's been difficult, and honestly heartbreaking, to see how it can affect children into adulthood. We're finally on the forefront of developing effective treatments, which is incredibly exciting and fulfilling."
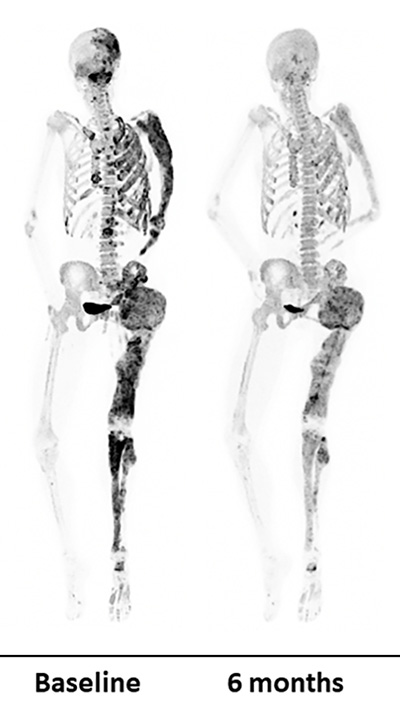
Dr. Boyce and her team recently put denosumab to the test in a clinical trial of eight adults with FD/MAS. The medicine markedly reduced abnormal bone turnover in the participants — an indication of improved bone quality and strength.
While the results were promising, abnormal bone turnover returned for all but one of the patients after they stopped taking the medication. The authors of the study, which was published as a correspondence report in the New England Journal of Medicine in April, noted that clinicians should take these effects into consideration when planning treatment for patients.
Dr. Boyce’s team is now testing whether early intervention with denosumab in children can prevent FD/MAS lesions from forming in the first place.
Future Directions
Dr. Collins and his collaborators have also worked to tackle FD/MAS from another angle by using small molecules to put the brakes on the overly active Gαs proteins. Although the effort has proven to be difficult so far, if successful, Dr. Collins said, such a therapy could be a “home run” for treating every aspect of FD/MAS all at once — the skin, endocrine, and skeletal symptoms. And because Gαs mutations are commonly found in cancers, these small molecules could also inform the development of new cancer therapies.
Looking into the future, NIDCR scientists believe that new and better treatments will become available for people with FD/MAS within the next decade. Advances in gene editing tools may help researchers create better stem cell models for studying FD/MAS. New biomaterials may improve surgical treatments for fractures and bone abnormalities that often characterize the disease.
Over the past 25 years, the patients and NIDCR researchers have become allies in a quest to understand a complex and variable disease. While there are still many questions to be answered, the researchers are committed to delivering solutions and hope to the patients and their families.
"It's very satisfying to take care of the patients and see them change, grow, and mature over the years," said Dr. Collins. "When a patient or family gives you a heartfelt ‘Thank you’, you know the research is worth it."
Related Links
- Symposium to Celebrate 25 Years of NIDCR FD/MAS Research
- Therapy for Rare Bone Disorder Shows Promise in NIH Clinical Trial
- Cracking Down on a Rare Bone Disorder
References:
- Bianco P, Kuznetsov SA, Riminucci M, Fisher LW, Spiegel AM, Robey PG. Reproduction of human fibrous dysplasia of bone in immunocompromised mice by transplanted mosaics of normal and Gsalpha-mutated skeletal progenitor cells. J Clin Invest. 1998 Apr 15;101(8):1737-44. doi: 10.1172/JCI2361.
- Boyce AM, Florenzano P, de Castro LF, Collins MT. Fibrous Dysplasia / McCune-Albright Syndrome. 2015 Feb 26 [Updated 2019 Jun 27]. In: Adam MP, Mirzaa GM, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023.
- de Castro LF, Burke AB, Wang HD, Tsai J, Florenzano P, Pan KS, et al. Activation of RANK/RANKL/OPG Pathway Is Involved in the Pathophysiology of Fibrous Dysplasia and Associated With Disease Burden. J Bone Miner Res. 2019 Feb;34(2):290-294. doi: 10.1002/jbmr.3602.
- de Castro LF, Michel Z, Pan K, Taylor J, Szymczuk V, Paravastu S, et al. Safety and Efficacy of Denosumab for Fibrous Dysplasia of Bone. N Engl J Med. 2023 Feb 23;388(8):766-768. DOI: 10.1056/NEJMc2214862.
- Lee JS, FitzGibbon E, Butman JA, Dufresne CR, Kushner H, Wientroub S, et al. Normal vision despite narrowing of the optic canal in fibrous dysplasia. N Engl J Med. 2002 Nov 21;347(21):1670-6. doi: 10.1056/NEJMoa020742.
- Szymczuk V, Taylor J, Boyce AM. Craniofacial Fibrous Dysplasia: Clinical and Therapeutic Implications. Curr Osteoporos Rep. 2023 Apr;21(2):147-153. doi: 10.1007/s11914-023-00779-6. Epub 2023 Feb 28.
Attention Editors
Reprint this article in your own publication or post to your website. NIDCR News articles are not copyrighted. Please acknowledge NIH's National Institute of Dental and Craniofacial Research as the source.
Subscribe for NIDCR Updates
Receive email updates about the latest advances in dental, oral, and craniofacial research.
November 2024